


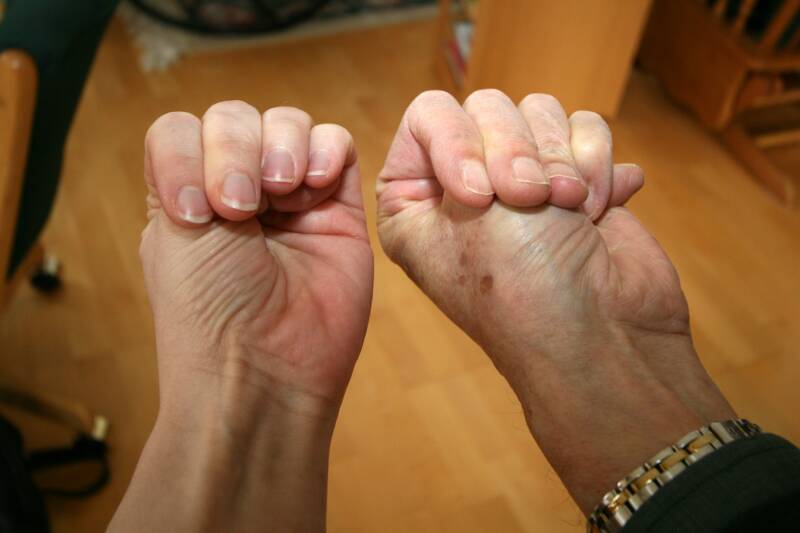

Alpha1-Antitrypsin Deficiency

A Cure For The Common Cold? New Drug Could Cure Nearly Any Viral Infection

Most bacterial infections can be treated with antibioticssuch as penicillin, discovered decades ago. However, such drugs are useless against viral infections, including influenza, the common cold, and deadly hemorrhagic fevers such as Ebola.
Now, in a development that could transform how viral infections are treated, a team of researchers at MIT's Lincoln Laboratory has designed a drug that can identify cells that have been infected by any type of virus, then kill those cells to terminate the infection.
In a paper published July 27 in the journal PLoS One, the researchers tested their drug against 15 viruses, and found it was effective against all of them - including rhinoviruses that cause the common cold, H1N1 influenza, a stomach virus, a poliovirus, dengue fever and several other types of hemorrhagic fever.read more...
What is your answer!

This young boy suffers from:
A) Hutchinson triad
B) Foeser-Kennedey syndrome
C) Marcus Gunn pupil
D) Marcus Gunn phenomenon
E) Argyll-Robertson pupil
What is the diagnose?
M.D. is a 35-year-old Malay labourer who presented
 BP136/85 mmHg, PR 98/min regular, non-collapsing
BP136/85 mmHg, PR 98/min regular, non-collapsing
with progressive shortness of breath on exertion 3/12.
There was no history of chest pain, palpitation, or
swelling of the legs. He sleeps with two pillows.
There was no history of acute rheumatic fever. He denies using illicit drugs. He was diagnosed to have a“murmur” in the heart in childhood.
Physical examination:
Afebrile
Orthopnea
Tachypnea
Polycythaemia
JVP: elevated
Heart: Apex beat 6th ICS 3 cm lateral to MCL,
forceful
No L parasterne
Loud split 2nd HS
EDM 1/6 in L sternal edge
EDM 1/6 in L sternal edge
Chest: NAD
Abdomen: Liver 2fb
CNS: NAD
What is your diagnosis?



Wilson disease
 What is Wilson disease?
What is Wilson disease?
Wilson disease is a genetic disorder that prevents the body from getting rid of extra copper. A small amount of copper obtained from food is needed to stay healthy, but too much copper is poisonous. In Wilson disease, copper builds up in the liver, brain, eyes, and other organs. Over time, high copper levels can cause life-threatening organ damage.
Who gets Wilson disease?
People who get Wilson disease inherit two abnormal copies of the ATP7B gene, one from each parent. Wilson disease carriers, who have only one copy of the abnormal gene, do not have symptoms. Most people with Wilson disease have no known family history of the disease. A person's chances of having Wilson disease increase if one or both parents have it.
About one in 40,000 people get Wilson disease. It equally affects men and women. Symptoms usually appear between ages 5 to 35, but new cases have been reported in people aged 2 to 72 years.
What causes Wilson disease?
Wilson disease is caused by a buildup of copper in the body. Normally, copper from the diet is filtered out by the liver and released into bile, which flows out of the body through the gastrointestinal tract. People who have Wilson disease cannot release copper from the liver at a normal rate, due to a mutation of the ATP7B gene. When the copper storage capacity of the liver is exceeded, copper is released into the bloodstream and travels to other organs—including the brain, kidneys, and eyes.
What are the symptoms of Wilson disease?
Wilson disease first attacks the liver, the central nervous system, or both.
A buildup of copper in the liver may cause ongoing liver disease. Rarely, acute liver failure occurs; most patients develop signs and symptoms that accompany chronic liver disease, including
- swelling of the liver or spleen
- jaundice, or yellowing of the skin and whites of the eyes
- fluid buildup in the legs or abdomen
- a tendency to bruise easily
- fatigue
A buildup of copper in the central nervous system may result in neurologic symptoms, including
- problems with speech, swallowing, or physical coordination
- tremors or uncontrolled movements
- muscle stiffness
- behavioral changes
Other signs and symptoms of Wilson disease include
- anemia
- low platelet or white blood cell count
- slower blood clotting, measured by a blood test
- high levels of amino acids, protein, uric acid, and carbohydrates in urine
- premature osteoporosis and arthritis
Kayser-Fleischer rings result from a buildup of copper in the eyes and are the most unique sign of Wilson disease. They appear in each eye as a rusty-brown ring around the edge of the iris and in the rim of the cornea. The iris is the colored part of the eye surrounding the pupil. The cornea is the transparent outer membrane that covers the eye.
How is Wilson disease diagnosed?
Wilson disease is diagnosed through a physical examination and laboratory tests.
During the physical examination, a doctor will look for visible signs of Wilson disease. A special light called a slit lamp is used to look for Kayser-Fleischer rings in the eyes. Kayser-Fleischer rings are present in almost all people with Wilson disease who show signs of neurologic damage but are present in only 50 percent of those with signs of liver damage alone.
Laboratory tests measure the amount of copper in the blood, urine, and liver tissue. Most people with Wilson disease will have a lower than normal level of copper in the blood and a lower level of corresponding ceruloplasmin, a protein that carries copper in the bloodstream. In cases of acute liver failure caused by Wilson disease, the level of blood copper is often higher than normal. A 24-hour urine collection will show increased copper in the urine in most patients who display symptoms. A liver biopsy—a procedure that removes a small piece of liver tissue—can show if the liver is retaining too much copper. The analysis of biopsied liver tissue with a microscope detects liver damage, which often shows a pattern unique to Wilson disease.
Genetic testing may help diagnose Wilson disease in some people, particularly those with a family history of the disease.
Wilson disease can be misdiagnosed because it is rare and its symptoms are similar to those of other conditions.
Who should be screened for Wilson disease?
Anyone with unexplained liver disease or neurologic symptoms with evidence of liver disease, such as abnormal liver tests and symptoms of liver disease, should be screened for Wilson disease. People with a family history of Wilson disease, especially those with an affected sibling or parent, should also be screened. A doctor can diagnose Wilson disease before the appearance of symptoms. Early treatment can reduce or even prevent illness.
How is Wilson disease treated?
Wilson disease requires lifelong treatment to reduce and control the amount of copper in the body.
Initial therapy includes the removal of excess copper, a reduction of copper intake, and the treatment of any liver or central nervous system damage.
The drugs d-penicillamine (Cuprimine) and trientine hydrochloride (Syprine) release copper from organs into the bloodstream. Most of the copper is then filtered out by the kidneys and excreted in urine. A potential major side effect of both drugs is that neurologic symptoms can become worse—a possible result of the newly released copper becoming reabsorbed by the central nervous system. About 20 to 30 percent of patients using d-penicillamine will also initially experience other reactions to the medication, including fever, rash, and other drug-related effects on the kidneys and bone marrow. The risk of drug reaction and neurologic worsening appears to be lower with trientine hydrochloride, which should be the first choice for the treatment of all symptomatic patients.
Pregnant women should take a lower dose of d-penicillamine or trientine hydrochloride during pregnancy to reduce the risk of birth defects. A lower dose will also help reduce the risk of slower wound healing if surgical procedures are performed during childbirth.
Zinc, administered as zinc salts such as zinc acetate (Galzin), blocks the digestive tract’s absorption of copper from food. Zinc removes copper too slowly to be used alone as an initial therapy for people who already have symptoms, but it is often used in combination with d-penicillamine or trientine hydrochloride. Zinc is safe to use at full dosage during pregnancy.
Maintenance therapy begins when symptoms improve and tests show that copper has been reduced to a safe level. Maintenance therapy typically includes taking zinc and low doses of either d-penicillamine or trientine hydrochloride. Blood and urine should be monitored by a health care provider to ensure treatment is keeping copper at a safe level.
People with Wilson disease should reduce their dietary copper intake. They should not eat shellfish or liver, as these foods may contain high levels of copper. Other foods high in copper—including mushrooms, nuts, and chocolate—should be avoided during initial therapy but, in most cases, may be eaten in moderation during maintenance therapy. People with Wilson disease should have their drinking water checked for copper content and should not take multivitamins that contain copper.
If the disorder is detected early and treated effectively, people with Wilson disease can enjoy good health.
Lingua Scrotalis

When grooves develop in the tongue, the condition is called scrotal tongue. It makes the tongue look wrinkled. Scrotal tongue has other names, too. It also is called furrowed tongue, lingua fissurata, lingua plicata, lingua scrotalis, plicated tongue or grooved tongue.
The tongue can have many grooves or a single groove down the middle with others branching off from it. Scrotal tongue affects 2% to 5% of the population of the United States. Rates in other countries vary. However, one study reported that the condition is found in up to 21% of people worldwide.
People usually are born with this condition, but may not notice it right away. It gets more noticeable as you age.
Sometimes, scrotal tongue occurs as a result of infection or poor nutrition. It also is seen in people with certain health conditions. These include:
- Melkersson-Rosenthal syndrome — This neurological disorder also includes facial nerve palsy and swelling of the face, especially the lips.
- Down syndrome
- Benign migratory glossitis, or geographic tongue — People with this condition have patches of smooth or bald tongue along with rougher areas where the taste buds are normal.
Symptoms
Scrotal tongue is a harmless condition. It usually has no symptoms. Sometimes, if the grooves are deep enough, you may get a burning sensation when you eat certain spicy or acidic foods. This is fairly rare, however. If the grooves are deep enough, they may harbor colonies of bacteria or bits of food. This can lead to bad breath or fungal infections of the tongue.
Diagnosis
Generally, people don't even know they have scrotal tongue until their dentist tells them. Your dentist or physician can diagnose this condition by looking at your tongue.
Expected Duration
Scrotal tongue is a lifelong condition.
Prevention
There is no way to prevent scrotal tongue.
Treatment
There is no treatment for scrotal tongue. If your tongue burns when you eat, you should avoid the foods that bother you. This should include cleaning your tongue daily with your toothbrush or a tongue scraper. You can buy these in drugstores.
When To Call A Professional
Check with your dentist if the grooves become uncomfortable.
Prognosis
Scrotal tongue is a lifelong condition, but it usually does not cause discomfort or problems.

Understanding Depression
 Feeling down from time to time is a normal part of life. But when emptiness and despair take hold and won't go away, it may be depression. More than just the temporary "blues," the lows of depression make it tough to function and enjoy life like you once did. Hobbies and friends don’t interest you like they used to; you’re exhausted all the time; and just getting through the day can be overwhelming. When you’re depressed, things may feel hopeless, but with help and support you can get better. But first, you need to understand depression. Learning about depression—including its signs, symptoms, causes, and treatment—is the first step to overcoming the problem.
Feeling down from time to time is a normal part of life. But when emptiness and despair take hold and won't go away, it may be depression. More than just the temporary "blues," the lows of depression make it tough to function and enjoy life like you once did. Hobbies and friends don’t interest you like they used to; you’re exhausted all the time; and just getting through the day can be overwhelming. When you’re depressed, things may feel hopeless, but with help and support you can get better. But first, you need to understand depression. Learning about depression—including its signs, symptoms, causes, and treatment—is the first step to overcoming the problem.What is depression?
We all go through ups and downs in our mood. Sadness is a normal reaction to life’s struggles, setbacks, and disappointments. Many people use the word “depression” to explain these kinds of feelings, but depression is much more than just sadness.
Some people describe depression as “living in a black hole” or having a feeling of impending doom. However, some depressed people don't feel sad at all—instead, they feel lifeless, empty, and apathetic.
Whatever the symptoms, depression is different from normal sadness in that it engulfs your day-to-day life, interfering with your ability to work, study, eat, sleep, and have fun. The feelings of helplessness, hopelessness, and worthlessness are intense and unrelenting, with little, if any, relief.
Are you depressed?
If you identify with several of the following signs and symptoms, and they just
won’t go away, you may be suffering from clinical depression.
Signs and symptoms of depressionwon’t go away, you may be suffering from clinical depression.
- you can’t sleep or you sleep too much
- you can’t concentrate or find that previously easy tasks are now difficult
- you feel hopeless and helpless
- you can’t control your negative thoughts, no matter how much you try
- you have lost your appetite or you can’t stop eating
- you are much more irritable and short-tempered than usual
- you have thoughts that life is not worth living (Seek help immediately if this is the case)
Depression varies from person to person, but there are some common signs and symptoms. It’s important to remember that these symptoms can be part of life’s normal lows. But the more symptoms you have, the stronger they are, and the longer they’ve lasted—the more likely it is that you’re dealing with depression. When these symptoms are overwhelming and disabling, that's when it's time to seek help.
Common signs and symptoms of depression
Feelings of helplessness and hopelessness. A bleak outlook—nothing will ever get better and there’s nothing you can do to improve your situation.
Loss of interest in daily activities. No interest in former hobbies, pastimes, social activities, or sex. You’ve lost your ability to feel joy and pleasure.
Appetite or weight changes. Significant weight loss or weight gain—a change of more than 5% of body weight in a month.
Sleep changes. Either insomnia, especially waking in the early hours of the morning, or oversleeping (also known as hypersomnia).
Irritability or restlessness. Feeling agitated, restless, or on edge. Your tolerance level is low; everything and everyone gets on your nerves.
Loss of energy. Feeling fatigued, sluggish, and physically drained. Your whole body may feel heavy, and even small tasks are exhausting or take longer to complete.
Self-loathing. Strong feelings of worthlessness or guilt. You harshly criticize yourself for perceived faults and mistakes.
Concentration problems. Trouble focusing, making decisions, or remembering things.
Unexplained aches and pains. An increase in physical complaints such as headaches, back pain, aching muscles, and stomach pain.
Classes of hypertension drugs

Diuretics
Diuretics ("water pills") increase the amount of sodium and water excreted into the urine by the kidneys. It is thought that they lower blood pressure mainly by reducing the volume of fluid in the blood vessels.
Diuretics commonly used for hypertension:
Acetazolamide - Diamox
Chlorthalidone - Thalitone
Hydrochlorothiazide - HydroDiuril, also sold as Microzide and Esidrix
Indapamide - Lozol
Metolazone - Zaroxolyn, also sold as Mykrox
Diuretics less commonly used for hypertension:
Amiloride hydrochloride - Midamor
Bumetanide - Bumex
Ethacrynic acid - Edecrin
Furosemide - Lasix
Spironolactone - Aldactone
Torsemide - Demadex
Triamterene - Dyrenium

Beta-blockers
Beta blockers block the effect of adrenaline on the cardiovascular system, slow the heart rate, and reduce stress on the heart and the arteries. Acebutolol - Sectral
Atenolol - Tenormin
Betaxolol - Kerlone
Bisoprolol - Zebeta, also sold as Ziac
Carteolol - Cartrol
Carvedilol - Coreg
Labetalol - Normodyne, also sold as Trandate
Metoprolol - Lopressor, also sold as Toprol
Nadolol - Corgard
Penbutolol - Levatol
Propranolol - Inderal, Inderal LA
Timolol - Blocadren
Acebutolol - Sectral
Atenolol - Tenormin
Betaxolol - Kerlone
Bisoprolol - Zebeta, also sold as Ziac
Carteolol - Cartrol
Carvedilol - Coreg
Labetalol - Normodyne, also sold as Trandate
Metoprolol - Lopressor, also sold as Toprol
Nadolol - Corgard
Penbutolol - Levatol
Propranolol - Inderal, Inderal LA
Timolol - Blocadren
Calcium Channel Blockers
Calcium channel blockers can reduce blood pressure by dilating the arteries and, in some cases, reducing the force of the heart's contractions.
Amlodipine - Norvasc, also sold as Caduet and Lotrel
Diltiazem - Cardizem, also sold as Dilacor and Tiazac
Felodipine - Plendil
Isradipine - DynaCirc
Nicardipine - Cardene
Nifedipine - Procardia XL, also sold as Adalat
Nisoldipine - Sular
Verapamil hydrochloride - Isoptin, also sold as Calan, Verelan, and Covera
Amlodipine - Norvasc, also sold as Caduet and Lotrel
Diltiazem - Cardizem, also sold as Dilacor and Tiazac
Felodipine - Plendil
Isradipine - DynaCirc
Nicardipine - Cardene
Nifedipine - Procardia XL, also sold as Adalat
Nisoldipine - Sular
Verapamil hydrochloride - Isoptin, also sold as Calan, Verelan, and Covera
Angiotensin Converting Enzyme Inhibitors
The angiotensin converting enzyme inhibitors (the "ACE inhibitors") can lower blood pressure by dilating the arteries. Benazepril - Lotensin
Captopril - Capoten
Enalapril - Vasotec, also sold as Vaseretic
Fosinopril - Monopril
Lisinopril - Prinivil, also sold as Zestril
Moexipril - Univasc
Quinapril - Accupril
Ramipril - Altace
Trandolapril - Mavik
Benazepril - Lotensin
Captopril - Capoten
Enalapril - Vasotec, also sold as Vaseretic
Fosinopril - Monopril
Lisinopril - Prinivil, also sold as Zestril
Moexipril - Univasc
Quinapril - Accupril
Ramipril - Altace
Trandolapril - Mavik
Angiotensin II Receptor Blockers
The angiotensin II receptor blockers (the "ARBs") also reduce blood pressure by dilating the arteries. Candesartan - Atacand
Irbesartan - Avapro
Losartan - Cozaar
Telmisartan - Micardis
Valsartan - Diovan
Candesartan - Atacand
Irbesartan - Avapro
Losartan - Cozaar
Telmisartan - Micardis
Valsartan - Diovan
Other, Less Commonly Used Hypertension Drugs
Clonidine - Catapres
Doxazosin - Cardura
Guanabenz - Wytensin
Guanfacine - Tenex
Hydralazine hydrochloride - Apresoline
Methyldopa - Aldomet
Prazosin - Minipress
Reserpine - Serpasil
Terazosin - Hytrin
Combination Drugs For Hypertension
Amiloride and hydrochlorothiazide - Moduretic
Amlodipine and benazepril - Lotrel
Atenolol and chlorthalidone - Tenoretic
Benazepril and hydrochlorothiazide - Lotensin HCT
Bisoprolol and hydrochlorothiazide - Ziac
Captopril and hydrochlorothiazide - Capozide
Enalapril and hydrochlorothiazide - Vaseretic
Felodipine and enalapril - Lexxel
Hydralazine and hydrochlorothiazide - Apresazide
Lisinopril and hydrochlorothiazide - Prinzide, also sold as Zestoretic
Losartan and hydrochlorothiazide - Hyzaar
Methyldopa and hydrochlorothiazide - Aldoril
Metoprolol and hydrochlorothiazide - Lopressor HCT
Nadolol and bendroflumethiazide - Corzide
Propranolol and hydrochlorothiazide - Inderide
Spironolactone and hydrochlorothiazide - Aldactazide
Triamterene and hydrochlorothiazide - Dyazide, also sold as Maxide
Verapamil extended release) and trandolapril - Tarka
Ehlers-Danlos Syndrome

Ehlers-Danlos syndrome occurs in both men and women of all ethnic backgrounds. This inherited disorder is caused by abnormalities in the production of collagen, a connective tissue found throughout the body. Ehlers-Danlos syndrome leads to defects in the inherent strength, elasticity, integrity, and healing properties of tissues.
Symptoms
Ehlers-Danlos syndrome is a group of disorders, including six major types classified according to their specific symptoms. Typical symptoms of EDS include:
- skin can be pulled away from the body as if elastic (skin extensibility)
- skin is fragile and tears easily (tissue fragility)
- joints can move beyond the normal range and easily dislocate (joint hypermobility)
Similar disorders
In some types of the syndrome, diagnosis can be made by a special test. In most cases, though, the syndrome is diagnosed based on the physical symptoms and family medical history.
However, there are several disorders that have some of the characteristics of Ehlers-Danlos syndrome. For example, in cutis laxa the skin is loose, hanging, and wrinkled. In cutis hyperelastica, the skin can be pulled away from the body but it does not tear. In Marfan syndrome, the joints are very mobile. In the past, Menkes disease, a copper metabolism disorder, was thought to be a form of Ehlers-Danlos syndrome. Because of these similar disorders, a correct diagnosis is very important.
Treatment
Although there is no cure, good medical care can provide a normal life span for most people with Ehlers-Danlos syndrome. Care focuses on:
- Preventing trauma to the joints. People with Ehlers-Danlos should not perform contortions or do heavy or repetitive lifting. Exercise and physical therapy are important to promote joint stability and reduce dislocations.
- Avoiding trauma to the skin and internal organs because of fragile skin and poor wound healing. This would include staying away from potentially traumatic recreational activities.
- Practicing meticulous dental care to avoid gum and tooth problems.
- Visiting the ophthalmologist regularly to be screened for eye problems.
- Genetic counseling, if available, especially for people wanting to have children.
Spot diagnosis

ANSWER
Cornu Cutaneum
A cutaneous horn (Cornu Cutaneum) is a relatively rare tumour, most often arising on sun-exposed skin in elderly men, usually after the Vth decade. The tumor is often conical, consisting of marked retention of stratum corneum. This lesion can be caused by many primary underlying processes that may be benign, premalignant or malignant, thus the importance of accurate determination of the nature of the conditions at the base of the lesion. Surgery is the treatment of choice. We describe an illustrative case with cutaneous horn of the forearm. We also review various known causes of cutaneous horn and the pertinent literature.

Arnold–Chiari malformation
 Arnold–Chiari malformation, or often simply Chiari malformation, is a malformation of the brain. It consists of a downward displacement of the cerebellar tonsils through the foramen magnum (the opening at the base of the skull), sometimes causing non-communicating hydrocephalus as a result of obstruction of cerebrospinal fluid (CSF) outflow. The cerebrospinal fluid outflow is caused by phase difference in outflow and influx of blood in the vasculature of the brain. It can cause headaches, fatigue, muscle weakness in the head and face, difficulty swallowing, dizziness, nausea, impaired coordination, and, in severe cases, paralysis.
Arnold–Chiari malformation, or often simply Chiari malformation, is a malformation of the brain. It consists of a downward displacement of the cerebellar tonsils through the foramen magnum (the opening at the base of the skull), sometimes causing non-communicating hydrocephalus as a result of obstruction of cerebrospinal fluid (CSF) outflow. The cerebrospinal fluid outflow is caused by phase difference in outflow and influx of blood in the vasculature of the brain. It can cause headaches, fatigue, muscle weakness in the head and face, difficulty swallowing, dizziness, nausea, impaired coordination, and, in severe cases, paralysis.Symptoms
- Headaches aggravated by Valsalva maneuvers, such as yawning, laughing, crying, coughing, sneezing or straining
- Tinnitus (ringing in the ears)
- Dizziness and vertigo
- Nausea
- Nystagmus (irregular eye movements)
- Facial pain
- Muscle weakness
- Impaired gag reflex
- Restless Leg Syndrome
- Sleep Apnea
- Dysphagia (difficulty swallowing)
- Impaired coordination
- Dysautonomia: tachycardia (rapid heart), syncope (fainting), polydipsia (extreme thirst), chronic fatigue
The blockage of Cerebro-Spinal Fluid (CSF) flow may also cause a syrinx to form, eventually leading to syringomyelia. Central cord symptoms such as hand weakness, dissociated sensory loss, and, in severe cases, paralysis may occur.
Diagnosis
Diagnosis is made through a combination of patient history, neurological examination, and Magnetic Resonance Imaging (MRI). The radiographic criteria for diagnosing a congenital Chiari I Malformation is a downward herniation of the cerebellar tonsils greater than 5 mm below the foramen magnum. Other imaging techniques involve the use of 3-D CT imaging of the brain and cine imaging (a movie of the brain) can be used to determine if the brainstem is being compressed by the pulsating arteries that surround it.
In the Syndrome of Occipitoatlantoaxial Hypermobility, cerebellar tonsillar herniation is typically only evident on an up-right MRI, due to the fact that the Chiari Malformation is gravitationally acquired by means of connective tissue weakness. 3-D CT imaging may aid in the diagnosis of related disorders such as retroflexed odontoid. Invasive cranial traction (lifting of the head off the spine) is often used as a confirmation of the diagnosis.
The diagnosis of a Chiari II Malformation can be made prenatally through Ultrasound.
Treatment
Once symptomatic onset occurs, a common treatment is decompression surgery, in which a neurosurgeon usually removes the lamina of the first and sometimes the second or even third cervical vertebrae and part of the occipital bone of the skull to relieve pressure. The flow of spinal fluid may be accompanied by a shunt. Since this surgery usually involves the opening of the dura mater and the expansion of the space beneath, a dural graft is usually applied to cover the expanded posterior fossa.
A small number of neurological surgeons believe that detethering the spinal cord as an alternate approach relieves the compression of the brain against the skull opening (foramen magnum), obviating the need for decompression surgery and associated trauma. However, this approach is significantly less documented in the medical literature, with reports on only a handful of patients. It should be noted that the alternative spinal surgery is also not without risk.
Parry–Romberg syndrome
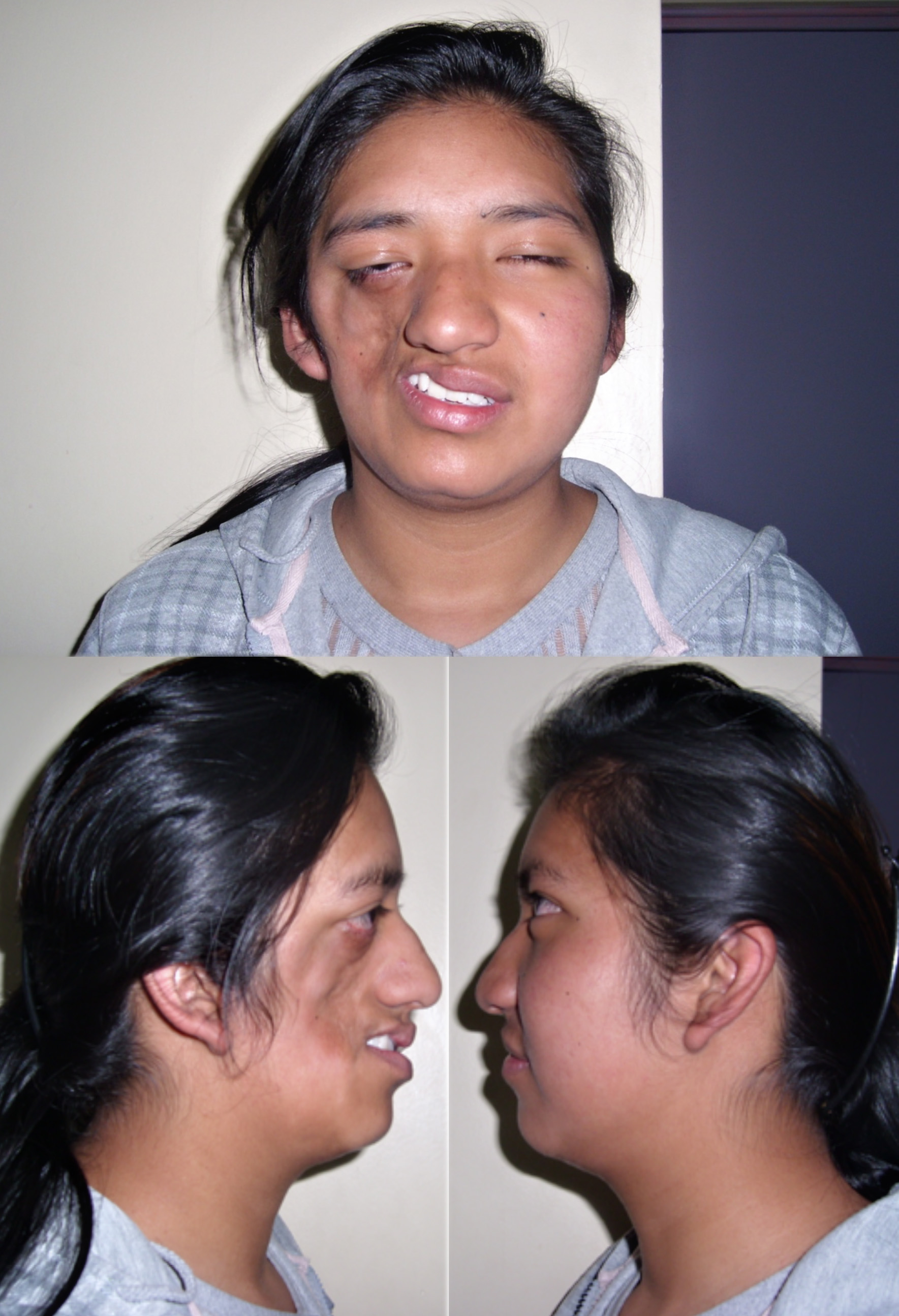
Parry–Romberg syndrome is a rare disorder characterized by slowly progressive degeneration (atrophy) of the soft tissues of half of the face (hemifacial atrophy). Some individuals may experience distinctive changes of the eyes and hair; and neurological abnormalities including episodes of uncontrolled electrical disturbances in the brain (seizures) and episodes of severe pain in tissues supplied by the fifth cranial nerve (trigeminal nerve) including the mouth, cheek, nose, and/or other facial tissues (trigeminal neuralgia). Symptoms and physical findings associated with Parry–Romberg syndrome usually become apparent during the first or early during the second decade of life. In rare cases, the disorder is apparent at birth. The majority of individuals with Parry–Romberg syndrome experience symptoms before the age of 20 years.
In affected individuals, initial facial changes usually involve the tissues above the upper jaw (maxilla) or between the nose and the upper corner of the lip (nasolabial fold) and progress to involve the angle of the mouth, the areas around the eye, the brow, the ear, and/or the neck. Progressive tissue wasting can be on either side of the face. In some rare cases, the atrophy may be bilateral. Affected areas may demonstrate shrinkage and atrophy of tissues beneath the skin (subcutaneous tissue), the layer of fat under the skin (subcutaneous fat), and underlying cartilage, muscle, and bone.
In addition, the skin overlying affected areas may become darkly pigmented (hyperpigmentation) with, in some cases, areas of hyperpigmentation and patches of unpigmented skin (vitiligo). Many individuals also experience atrophy of half of the upper lip and tongue as well as abnormal exposure, delayed eruption, or wasting of the roots of certain teeth on the affected side. Symptoms of Parry–Romberg syndrome may begin at any age. Facial atrophy may cease abruptly, or progress slowly and then become stationary. If the atrophy becomes stationary, it may reactivate later in life. In other cases, the atrophy may progress indefinitely. In some cases, hair abnormalities may also appear on the affected side, including whitening (blanching) of the hair as well as abnormal bald patches on the scalp and loss of eyelashes and the middle (median) portion of the eyebrows (alopecia).
In addition, some individuals with Parry–Romberg syndrome may also experience associated neurological abnormalities. These may include severe headaches that last for extended periods of time and may be accompanied by visual abnormalities, nausea, and vomiting (migraines); facial pain (trigeminal neuralgia); and/or periods of uncontrolled electrical disturbances in the brain (seizures) that usually are characterized by rapid spasms of a muscle group that spread to adjacent muscles (contralateral Jacksonian epilepsy). The range and severity of associated symptoms and findings may vary from case to case. In most cases, Parry–Romberg syndrome appears to occur randomly for unknown reasons (sporadically)
Case 3
A 17-year-old boy comes to the physician
because of right groin pain for 2 hours;
elevation of the scrotum does not relieve the
pain. There is no history of trauma. He is
sexually active and has had multiple sexual
partners over the past 3 years. He does not use
condoms regularly. He had chlamydial
urethritis 1 year ago treated with doxycycline.
Examination shows an enlarged, swollen,
erythematous, and acutely tender right
scrotum. The left testis is found in a horizontal
position; the cremasteric reflex is absent on the
right. Which of the following is the most likely
diagnosis?
(A) Epididymitis
(B) Hemorrhagic tumor
(C) Incarcerated hernia
(D) Torsion of the testicle
(E) Torsion of the testicular appendix
because of right groin pain for 2 hours;
elevation of the scrotum does not relieve the
pain. There is no history of trauma. He is
sexually active and has had multiple sexual
partners over the past 3 years. He does not use
condoms regularly. He had chlamydial
urethritis 1 year ago treated with doxycycline.
Examination shows an enlarged, swollen,
erythematous, and acutely tender right
scrotum. The left testis is found in a horizontal
position; the cremasteric reflex is absent on the
right. Which of the following is the most likely
diagnosis?
(A) Epididymitis
(B) Hemorrhagic tumor
(C) Incarcerated hernia
(D) Torsion of the testicle
(E) Torsion of the testicular appendix

Spot diagnosis

ANSWER
Dermatitis herpetiformis
Dermatitis herpetiformis (DH) is an autoimmune blistering disorder associated with a gluten-sensitive enteropathy (GSE). Dermatitis herpetiformis is characterized by grouped excoriations; erythematous, urticarial plaques; and papules with vesicles. The classic location for dermatitis herpetiformis lesions is on the extensor surfaces of the elbows, knees, buttocks, and back. Dermatitis herpetiformis is exquisitely pruritic, and the vesicles are often excoriated to erosions by the time of physical examination.
Pathophysiology
Dermatitis herpetiformis is a disease of the skin caused by the deposition of IgA in the papillary dermis, which triggers an immunologic cascade, resulting in neutrophil recruitment and complement activation. Dermatitis herpetiformis is the result of an immunologic response to chronic stimulation of the gut mucosa by dietary gluten.
Pathophysiology
Dermatitis herpetiformis is a disease of the skin caused by the deposition of IgA in the papillary dermis, which triggers an immunologic cascade, resulting in neutrophil recruitment and complement activation. Dermatitis herpetiformis is the result of an immunologic response to chronic stimulation of the gut mucosa by dietary gluten.

What is diagnosis?

nausea, vomiting, and increasingly severe abdominal pain that radiates to his left shoulder and back.
He appears extremely dehydrated and is short of breath. His temperature is 37.8°C (100°F), pulse is 120/min,
respirations are 18/min, and blood pressure is 80/60 mm Hg. Abdominal examination shows distention with
epigastric tenderness. Bowel sounds are decreased. Rectal examination shows no abnormalities. Test of the
stool for occult blood is negative. Laboratory studies show:
Hemoglobin 5.5 g/dL
Leukocyte count 15,500/mm3
Serum
Ca2+ 7.5 mg/dL
Amylase 750 U/L
A CT scan of the abdomen is shown.
Tree Man

Half Man Half Tree / Tree Man
32 year old Dede Kosawa, also known as 'Tree Man', is one of the world's most extraordinary people. He lives in a remote village in Indonesia with his two children, trying to care for them. Dede, a former fisherman, has an incredible skin condition: he has root like structures growing out of his body - branches that can grow up to 5cm a year and which protrude from his hands and feet, and welts covering his whole body.
He is known locally as ‘Tree Man’ and his condition has baffled local doctors for 20 years. In an attempt to earn a living to support his family, he is part of a circus troupe, displaying his 'Tree Man' limbs along with others afflicted with skin deformities in ‘freak’ shows.Mermaid syndrome
 Mermaid syndrome or sirenomelia is a condition where the legs of a new-born child are fused together. The child may have two feet sticking out to the sides like flippers or no feet at all. It is the former that gives sirenomelia its name. The bones of the feet and legs may be missing entirely or fused.
Mermaid syndrome or sirenomelia is a condition where the legs of a new-born child are fused together. The child may have two feet sticking out to the sides like flippers or no feet at all. It is the former that gives sirenomelia its name. The bones of the feet and legs may be missing entirely or fused.Usually, children with mermaid syndrome die shortly after birth. The condition occurs along with other abnormalities that affect the kidneys, bladder, genitals and rectum. The lungs may also be affected. With such an array of problems it is no wonder that survival is so rare.
The cause of mermaid syndrome is not known for sure. It is generally related to poor prenatal care or illness. Genetic predisposition is also a possibility. The condition can be detected by sonogram and the recommended treatment, given the poor prognosis, is termination of the pregnancy.
There have been three cases of children with mermaid syndrome surviving. The most recent was a girl in Peru named Milagros Cerron. Her condition was limited to the skin and blood vessels of her legs - otherwise the bones and muscles were present and her legs could move independently under her skin. Other internal organ were also affected especially the kidneys. Fortunately, Milagros is mentally a normal child.
After an operation in June, 2005, Milagros' legs were separated. The doctors commented that the surgery had gone far better than anticipated. She will need corrective surgery for her internal organs for the next 10 to 15 years but the prognosis is excellent. As of this year, 2006, she has begun to walk and doctors say she should be walking normally by the end of the year.
An unusual presentation of brucellosis
A 26-year-old man referred to our center (Alzahra University Hospital, Isfahan, Iran) with complaints of fever, ataxia and dysarthria. His problem began 3 months prior to the recent hospitalization with low grade fever, chilly sensation, cold sweating, loss of appetite and 10 Kg weight loss. He was also complaining from arthralgia in different joints and could not walk without help. His dysarthria and vomiting started 10 and 2 days before the admission, respectively. He had undergone left nephrectomy for congenital urethropelvic junction obstruction and severe hydronephrosis 2 years ago. He was symptom free up to the recent referral.
Physical examination revealed an ill looking young man with body temperature of 39°C, pulse rate 45/Min, and blood pressure of 110/60 mm Hg. He was dysarthteric and truncal ataxia was also observed. On auscultation he had muffled heart sounds without any murmur. The abdomen was soft with tender hepatomegaly 3 cm below the costal margin.
Results of laboratory tests made on admission were as follow: White blood cell count: 17300/mm-3 with 91%Neutrophils, Patelet count: 89000/mm-3, Hemoglobin: 14.1 g/dl, C-reactive protein: 15 mg/dl, Erythrocyte sedimentation rate: 41 mm/h, Blood urea nitrogen(BUN): 40 mg/dL, Creatine(Cr): 3.9 mg/dL, SGOT: 338 mg/dL, SGPT: 151 mg/dL, Alkaline phosphatase: 593 mg/dL, Creatine phosphokinase(CPK): 468 mg/dL, CPK(MB): 38 mg/dL,. Urinalysis revealed moderate hematuria and 24-hour urinalysis (U.A) revealed proteinuria (1143 mg/dL). ANA, ANCA, anti-dsDNA, RF, Anti Cardiolopin Ab, Lupus Anticoagulant were all negative. A PPD skin test was negative.
Chest X ray was normal. Electrocardiogram showed sinus bradycardia and high T waves in the precordial leads. Echocardiographic examination revealed septal hypokinesia and ejection fraction of 30%. No vegetation was reported. Brain MRI showed white matter changes in corpus callosum, periventricular area and centrum semiovalis (Fig. 1). Liver and kidney biopsies as well as lumbar puncture were not performed due to our patient's refusal.

Physical examination revealed an ill looking young man with body temperature of 39°C, pulse rate 45/Min, and blood pressure of 110/60 mm Hg. He was dysarthteric and truncal ataxia was also observed. On auscultation he had muffled heart sounds without any murmur. The abdomen was soft with tender hepatomegaly 3 cm below the costal margin.
Results of laboratory tests made on admission were as follow: White blood cell count: 17300/mm-3 with 91%Neutrophils, Patelet count: 89000/mm-3, Hemoglobin: 14.1 g/dl, C-reactive protein: 15 mg/dl, Erythrocyte sedimentation rate: 41 mm/h, Blood urea nitrogen(BUN): 40 mg/dL, Creatine(Cr): 3.9 mg/dL, SGOT: 338 mg/dL, SGPT: 151 mg/dL, Alkaline phosphatase: 593 mg/dL, Creatine phosphokinase(CPK): 468 mg/dL, CPK(MB): 38 mg/dL,. Urinalysis revealed moderate hematuria and 24-hour urinalysis (U.A) revealed proteinuria (1143 mg/dL). ANA, ANCA, anti-dsDNA, RF, Anti Cardiolopin Ab, Lupus Anticoagulant were all negative. A PPD skin test was negative.
Chest X ray was normal. Electrocardiogram showed sinus bradycardia and high T waves in the precordial leads. Echocardiographic examination revealed septal hypokinesia and ejection fraction of 30%. No vegetation was reported. Brain MRI showed white matter changes in corpus callosum, periventricular area and centrum semiovalis (Fig. 1). Liver and kidney biopsies as well as lumbar puncture were not performed due to our patient's refusal.

Since he was a shepherd from an endemic area for brucellosis and gave a history about the consumption of non-pasteurized dairy products, standard tube agglutination test (STAT), 2-mercaptoethanol (2-ME), and Coombs test were performed (Antigen from the Pasteur Institute of Iran, Tehran). STAT and 2-ME titers were 1:80 and 1:40 respectively. Titer of Coombs test was 1:80. Enzyme-linked immunosorbent assay (IBL, Germany) for IgG and IgM was negative. Blood culture was performed once and it was positive for brucellosis. The Brucella omp2a gene was used as target DNA for PCR amplification. Polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) [5] demonstrated the presence of Brucella mellitensis bv 1. Doxycycline (2 × 100 mg/day p.o.) and Rifampicin (1 × 600 mg/day p.o.) were started. The patient received this treatment for six weeks. One week after the initiation of the treatment our patient's symptoms subsided and he became afebrile. After 2 weeks he had normal liver function tests, BUN, and Cr levels and transthoracic echocardiography was normal too. The patient was followed-up for 10 months. In 6th months he experienced a relapse of brucellosis with signs and symptoms similar to the previous episode of the infection. The standard treatment regimen was started again. He is now receiving this treatment and is asymptomatic but still dysarthric. Our plan is to continue the medication for at least 6 months.
Interesting case
A 25-year-old woman with no clinically significant medical history went bungee jumping from a vertical height of 150 ft (45.7 m). Immediately afterward, she noticed a substantial decrease in vision in her left eye, with a large central scotoma; no other symptoms were noted and there was no pain… the foveal and macular architecture was obscured by a large macular hemorrhage just below the level of the internal limiting membrane (Panel B, arrow).


What is diagnosis
A 65-year-old male with a past history of cigarette smoking and asbestos exposure (in the 1970s) presented to the emergency department with a one-month history of progressive dyspnea, right-sided pleuritic chest pain, cough productive of white-coloured sputum and malaise. His health problems had commenced four months before presentation while he was vacationing at a northern Ontario resort. At that time, he had felt unwell and had developed a fever with rightsided pleuritic chest pain that radiated to his right shoulder. The diagnosis was an upper respiratory tract infection, made by the local physician; the patient was treated with a 10-day course of cephalexin. Although his condition had initially improved after the antibiotic therapy, during the month before presentation he had experienced increasing fatigue, cough with clear sputum production and a loss of appetite. He also developed worsening right-sided pleuritic chest pain that radiated to the right shoulder, dyspnea and orthopnea. He had no nausea, vomiting, diarrhea or hemoptysis. However, he had lost 4 kg and had drenching night sweats over the previous three and a half months. Further history revealed that he had drunk well water during his vacation in northern Ontario and that several families who were with him at that time also became ill, although he was not aware of the nature of their symptoms.
On examination, he was a thin male in some respiratory distress and was afebrile. His blood pressure was 140/90 mmHg, and he had a heart rate of 80 beats/min and a respiratory rate of 30 breaths/min. Chest examination revealed percussion dullness, decreased breath sounds, crackles, and bronchial breathing in the right base and mid-lung fields. No clubbing was observed. There was no jugular venous distention, and heart sounds were normal without any murmurs. His abdomen was flat, but he had mild right upper quadrant tenderness on deep palpation. No abdominal masses were palpable, no organomegaly was appreciated and there were no stigmata of chronic liver disease. Bowel sounds were present.
The patient's oxygen saturation was 89% on room air. The white blood cell count revealed mild leukocytosis of 12.2x109/L without a left shift, but hemoglobin and platelet counts were normal. The erythrocyte sedimentation rate was 38 mm/h. The level of serum aspartate aminotransferase was 28 U/L, while the alanine aminotransferase level was mildly elevated at 47 U/L; the alkaline phosphatase and gammaglutamyl transpeptidase levels were moderately elevated at 380 U/L and 243 U/L, respectively. The patient's total bilirubin concentration was normal, but the albumin concentration was depressed at 32 g/L. All other serum chemistry values were within normal limits.
A chest x-ray (Figure 1) showed a large right pleural effusion with associated
consolidation or subsegmental atelectasis in the right middle and lower lung zones. An underlying parenchymal mass could not be excluded. Computed tomography of the thorax with contrast enhancement (Figure 2) revealed no masses in the lung
parenchyma or the mediastinum. However, it demonstrated a 10 cm fluid collection occupying most of the right lobe of the liver, with peripheral enhancement consistent with a massive liver abscess.
Figure 1.

Figure 2

Percutaneous computed tomography-guided drainage of the liver abscess ensued, and 200 mL of bloody purulent fluid was aspirated. In addition, a chest tube was inserted, and 900 mL of fluid of similar consistency with numerous leukocytes was removed from the thorax. Microscopic examination of the fluids did not reveal any microorganisms, and cultures of the fluids and blood were negative for bacteria. Thereafter, therapy consisting of intravenous clindamycin 600 mg every 8 h and oral ciprofloxacin 750 mg bid was initiated.
What is your diagnosis?
On examination, he was a thin male in some respiratory distress and was afebrile. His blood pressure was 140/90 mmHg, and he had a heart rate of 80 beats/min and a respiratory rate of 30 breaths/min. Chest examination revealed percussion dullness, decreased breath sounds, crackles, and bronchial breathing in the right base and mid-lung fields. No clubbing was observed. There was no jugular venous distention, and heart sounds were normal without any murmurs. His abdomen was flat, but he had mild right upper quadrant tenderness on deep palpation. No abdominal masses were palpable, no organomegaly was appreciated and there were no stigmata of chronic liver disease. Bowel sounds were present.
The patient's oxygen saturation was 89% on room air. The white blood cell count revealed mild leukocytosis of 12.2x109/L without a left shift, but hemoglobin and platelet counts were normal. The erythrocyte sedimentation rate was 38 mm/h. The level of serum aspartate aminotransferase was 28 U/L, while the alanine aminotransferase level was mildly elevated at 47 U/L; the alkaline phosphatase and gammaglutamyl transpeptidase levels were moderately elevated at 380 U/L and 243 U/L, respectively. The patient's total bilirubin concentration was normal, but the albumin concentration was depressed at 32 g/L. All other serum chemistry values were within normal limits.
A chest x-ray (Figure 1) showed a large right pleural effusion with associated
consolidation or subsegmental atelectasis in the right middle and lower lung zones. An underlying parenchymal mass could not be excluded. Computed tomography of the thorax with contrast enhancement (Figure 2) revealed no masses in the lung
parenchyma or the mediastinum. However, it demonstrated a 10 cm fluid collection occupying most of the right lobe of the liver, with peripheral enhancement consistent with a massive liver abscess.
Figure 1.

Figure 2

Percutaneous computed tomography-guided drainage of the liver abscess ensued, and 200 mL of bloody purulent fluid was aspirated. In addition, a chest tube was inserted, and 900 mL of fluid of similar consistency with numerous leukocytes was removed from the thorax. Microscopic examination of the fluids did not reveal any microorganisms, and cultures of the fluids and blood were negative for bacteria. Thereafter, therapy consisting of intravenous clindamycin 600 mg every 8 h and oral ciprofloxacin 750 mg bid was initiated.
What is your diagnosis?
Subscribe to:
Comments (Atom)
About




LinkWithin
